



Beyond SAR: How Scaffold Hopping Enables First-in-Class Drug Discovery
Scaffold hopping and bioisosteric design expand chemical space beyond traditional SAR, enabling novel, first-in-class drug discovery.
Introduction
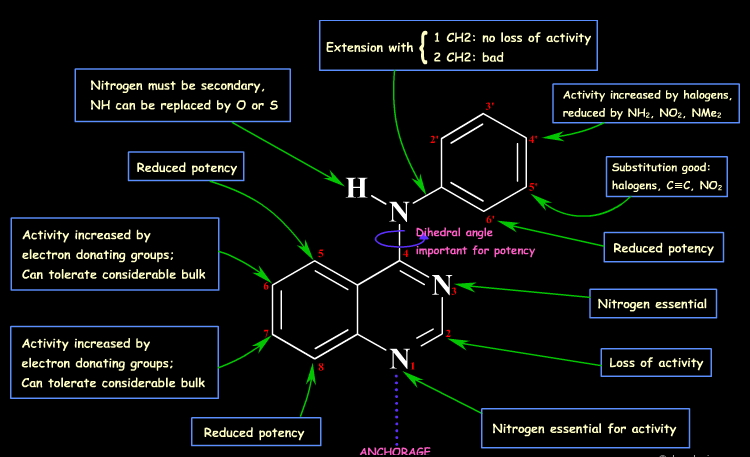
Drug discovery rarely progresses through a perfectly linear path. Early hits emerging from screening campaigns may demonstrate promising biological activity, but transforming those molecules into viable drug candidates requires extensive chemical optimization. Traditionally, medicinal chemists rely on structure–activity relationship (SAR) studies to refine molecules and improve potency, selectivity, and pharmacokinetic properties.
SAR remains a foundational principle in medicinal chemistry. However, as drug discovery increasingly targets complex biological systems, relying solely on incremental scaffold modifications often limits innovation. In many discovery programs, the real breakthroughs occur when researchers move beyond the original chemical scaffold.
This is where scaffold hopping and bioisosteric design play a crucial role. At Medvolt, these strategies are integrated into AI-driven molecular design workflows within the MedGraph platform, enabling discovery teams to systematically explore alternative chemotypes while preserving the key interactions responsible for biological activity.
The Limits of Traditional SAR
Structure–activity relationship analysis involves systematically modifying substituents around a core molecular scaffold and measuring how these changes influence biological activity. This iterative approach has enabled the development of many successful medicines by improving properties such as target affinity, metabolic stability, and safety.
However, SAR optimization typically explores only local chemical space. Most modifications occur around the same central scaffold, which limits the structural diversity that can be achieved.
This creates several challenges:
- Chemical redundancy where many analogs share similar liabilities
- Limited novelty from an intellectual property perspective
- Biological ceilings where potency cannot be improved further
- Persistent physicochemical issues that SAR alone cannot resolve
In modern computational drug discovery, platforms like MedGraph help address these limitations by analyzing protein–ligand interaction networks and identifying opportunities to explore alternative scaffolds and bioisosteric replacements while preserving key pharmacophoric features.
Rather than extending SAR indefinitely, researchers can strategically transition to scaffold diversification, unlocking new regions of chemical space.
What Is Scaffold Hopping?
Scaffold hopping is a medicinal chemistry strategy in which the central molecular framework of a compound is replaced with a structurally distinct core while maintaining the spatial arrangement of key pharmacophoric elements.
Unlike conventional SAR modifications that adjust peripheral substituents, scaffold hopping transforms the underlying architecture of the molecule. The objective is to retain the interactions necessary for biological activity while introducing a new chemotype that may offer improved drug-like properties.
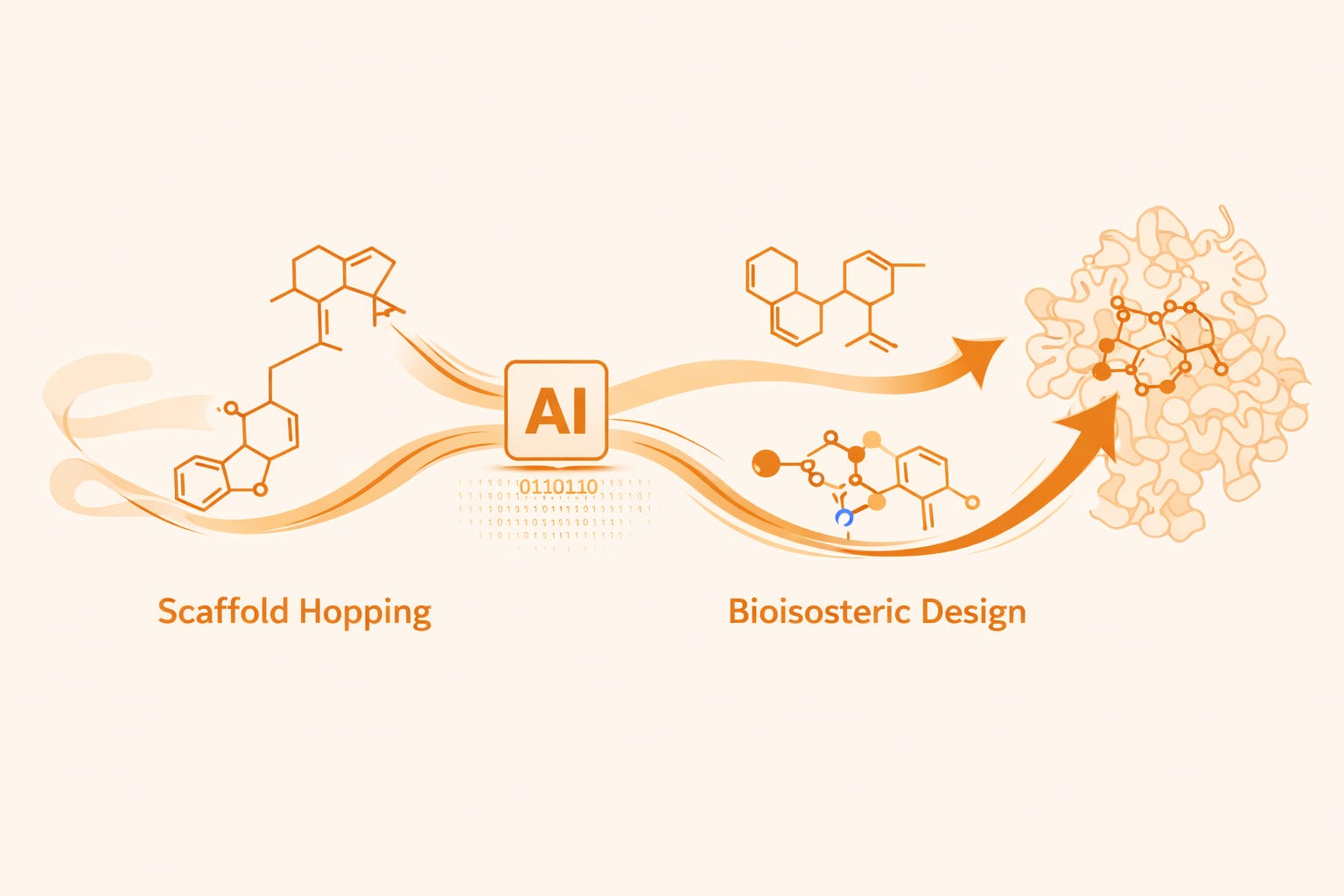
This strategy enables researchers to:
- Discover alternative chemotypes targeting the same binding pocket
- Improve pharmacokinetic or physicochemical properties
- Overcome resistance mechanisms
- Generate novel intellectual property
In computational workflows used at Medvolt, scaffold hopping is guided by interaction fingerprint analysis and pharmacophore mapping. By identifying the residues and interaction patterns responsible for binding, MedGraph can propose alternative scaffolds that preserve those interactions even when the chemical core changes.
This approach ensures that structural innovation does not compromise biological relevance.
Bioisosteric Design: Preserving Function While Changing Chemistry
Closely related to scaffold hopping is bioisosteric replacement, a strategy in which atoms or functional groups are substituted with chemically similar alternatives that maintain biological activity.
Bioisosteric modifications help medicinal chemists adjust molecular properties while preserving key interactions with the biological target. Classic examples include replacing phenyl rings with heteroaromatic systems or substituting hydrogen atoms with fluorine to influence metabolic stability.
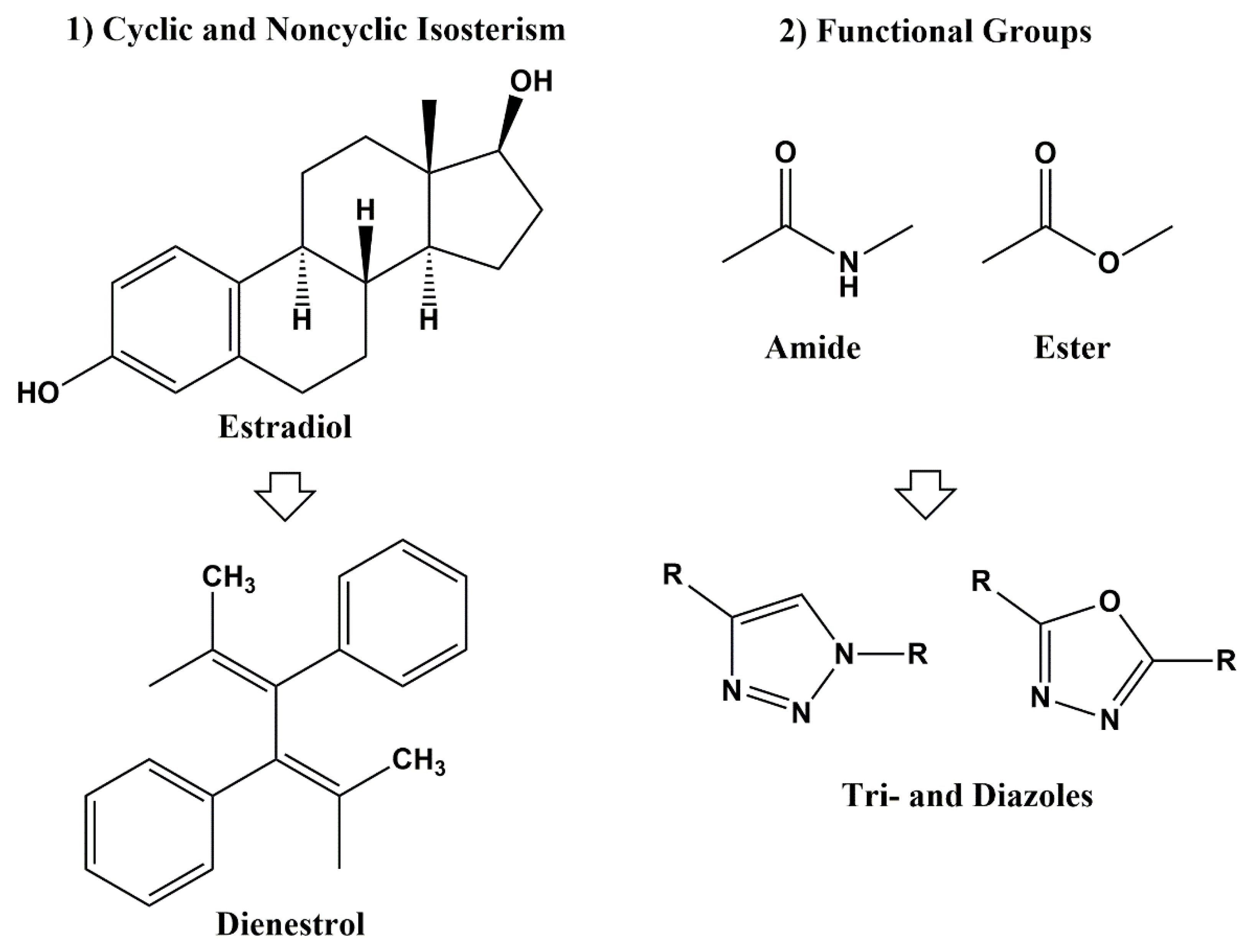
Strategic bioisosteric design can improve:
- Binding affinity and orientation
- Metabolic stability
- Solubility and permeability
- Toxicity profiles
When integrated with computational analysis of binding interactions, bioisosteric replacements become a powerful optimization tool. AI-driven platforms can rapidly evaluate multiple replacement possibilities while ensuring that pharmacophoric alignment and interaction networks remain intact.
In MedGraph-driven workflows, bioisosteric replacements are evaluated alongside scaffold hopping proposals. This enables discovery teams to identify structural modifications that simultaneously improve potency, selectivity, and developability while maintaining key binding interactions.
Such AI-assisted design significantly accelerates the exploration of viable chemical alternatives.
Why Scaffold Hopping Is Essential for First-in-Class Discovery
Scaffold hopping becomes particularly valuable when pursuing first-in-class therapeutics against novel biological targets.
In these cases, early hits often represent imperfect starting points. They may demonstrate biological activity but possess chemical features that limit their optimization potential. Without exploring alternative scaffolds, discovery programs risk being trapped within suboptimal chemical frameworks.
By introducing structurally distinct cores that preserve interaction geometry, scaffold hopping enables researchers to uncover entirely new classes of molecules capable of targeting the same biological site.
Within Medvolt’s discovery workflows, interaction analysis within MedGraph helps identify critical binding motifs that must be preserved during scaffold replacement. This allows scaffold hopping to be guided by biological insight rather than purely structural similarity, increasing the probability of discovering viable new chemical series.
The Challenge of Identifying Alternative Scaffolds
Although scaffold hopping offers significant advantages, identifying viable alternative scaffolds is challenging. The number of possible molecular frameworks that maintain pharmacophoric alignment is extremely large.
Historically, medicinal chemists relied heavily on intuition, literature precedents, and manual chemical exploration to identify scaffold replacements. While effective, these approaches struggle to scale with the enormous size of modern chemical space.
Artificial intelligence is now transforming this process. Machine learning models trained on chemical and structural datasets can identify scaffold transformations that preserve key interaction motifs. These models can also evaluate bioisosteric replacements that improve physicochemical properties while maintaining biological function.
By integrating generative chemistry with structure-based modeling, AI-assisted systems allow researchers to systematically explore scaffold alternatives rather than relying solely on human intuition.
Platforms like MedGraph apply these approaches to analyze large chemical spaces and identify alternative chemotypes that medicinal chemists might not discover through traditional exploration.
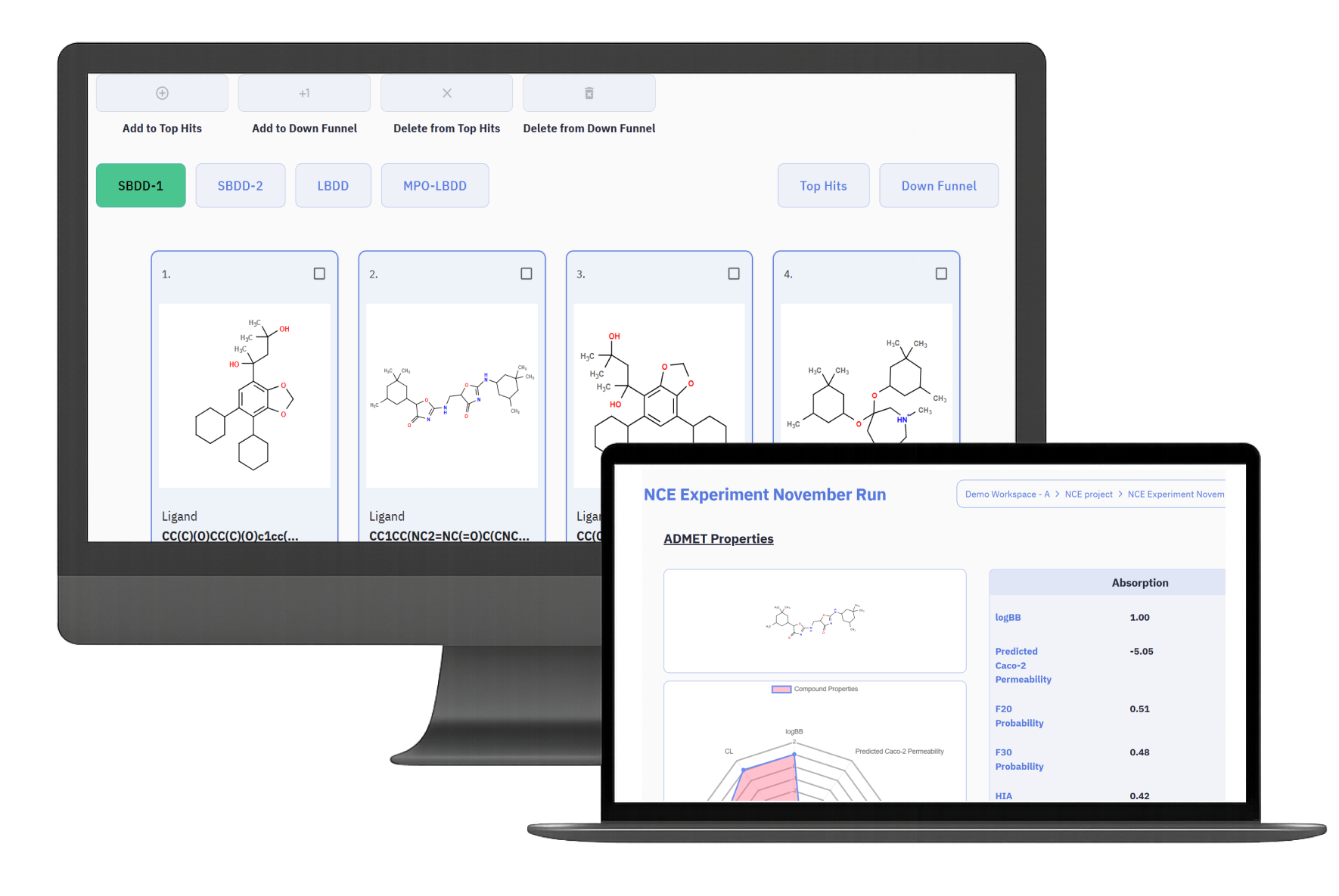
AI-Driven Scaffold Exploration in Modern Drug Discovery
The integration of AI and computational chemistry has enabled scaffold hopping to become a scalable discovery strategy. Modern molecular design platforms combine several computational approaches to generate and evaluate scaffold alternatives.
These include:
- Pharmacophore and interaction fingerprint analysis
- Generative models for proposing new chemotypes
- Structure-based docking and interaction evaluation
- Bioisosteric replacement libraries
- Multi-parameter optimization algorithms
By combining these methods, discovery teams can rapidly explore chemical space and identify molecules that balance potency, selectivity, novelty, and developability.
Such AI-assisted approaches help ensure that scaffold hopping does not merely produce structurally novel molecules, but also yields candidates that retain strong biological relevance.
Within Medvolt’s MedGraph platform, these methods are combined to support AI-assisted scaffold diversification. By simultaneously considering interaction geometry, physicochemical properties, and biological context, MedGraph can propose scaffold alternatives that balance novelty with functional relevance.
Medvolt’s Approach to Scaffold Hopping and Bioisosteric Design
At Medvolt, scaffold hopping and bioisosteric design are integrated into AI-driven drug discovery workflows that combine chemical intelligence with structural modeling.
Medvolt’s platform enables the AI-assisted identification of alternative scaffolds and bioisosteric replacements that preserve critical protein–ligand interactions while improving potency, selectivity, novelty, and developability.
These workflows involve:
- Mapping interaction patterns between ligands and target proteins
- Identifying pharmacophoric features responsible for binding
- Generating alternative chemotypes that preserve interaction geometry
- Evaluating bioisosteric substitutions to optimize molecular properties
- Integrating structure-based validation methods to ensure biological relevance
Because scaffold hopping is rarely a standalone step, these capabilities are embedded within broader discovery pipelines that integrate virtual screening, molecular modeling, and predictive simulations. This modular approach allows discovery teams to move seamlessly from hit identification to lead optimization while exploring diverse chemical architectures.
Looking Beyond SAR
Structure–activity relationship studies will continue to play a central role in medicinal chemistry. However, the increasing complexity of biological targets demands strategies that extend beyond incremental modification.
Scaffold hopping and bioisosteric design provide a pathway for exploring new chemical space while preserving biological activity. When combined with AI-driven molecular design and structural modeling, these approaches allow drug discovery programs to move beyond the limitations of traditional SAR.
By combining AI-driven design strategies with structural and physicochemical analysis, platforms like MedGraph enable a more systematic exploration of chemical diversity.
As computational tools continue to evolve, the ability to intelligently navigate chemical space will become a defining factor in identifying first-in-class therapeutic candidates capable of addressing some of the most challenging diseases.
