



Docking vs Molecular Dynamics: What’s the Difference?
Docking and molecular dynamics serve different roles in drug discovery, combining speed, scale, and realism for better binding predictions.
Introduction
In modern drug discovery, computational methods play a central role in identifying and optimizing potential drug candidates.
Among these, two techniques are widely used:
- molecular docking
- molecular dynamics (MD) simulations
They are often mentioned together.
Sometimes even used interchangeably.
But in reality, they answer very different questions.
Understanding the distinction between docking and molecular dynamics is not just a technical detail.
It is fundamental to making better decisions in drug discovery.
The Starting Point: What Are We Trying to Predict?
At the core of small molecule drug discovery is a simple objective:
Will this molecule bind to the target protein, and how well?
But binding is not a single event.
It involves:
- orientation
- interactions
- flexibility
- environmental effects
- time-dependent behavior
Docking and molecular dynamics approach this problem from two different perspectives.
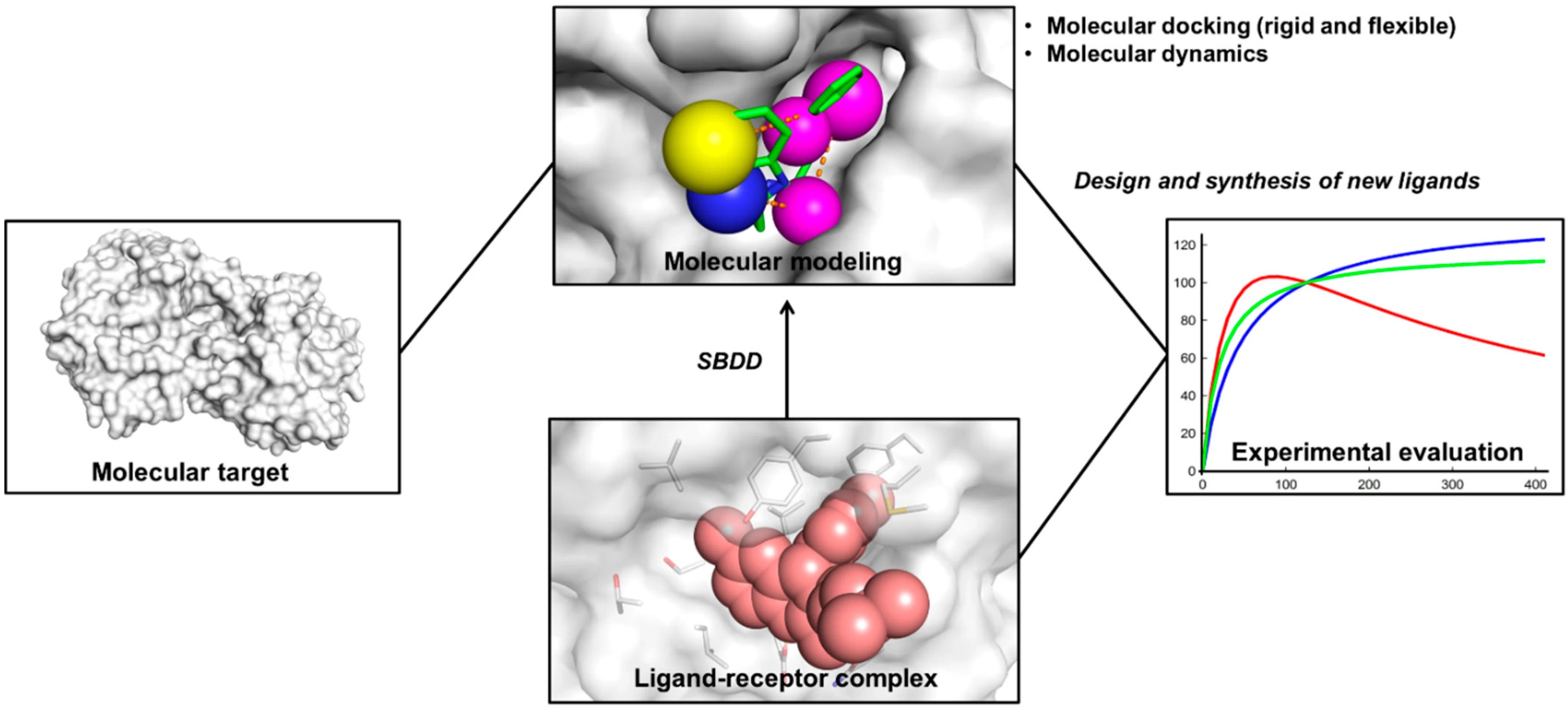
What Is Molecular Docking?
Molecular docking is a computational technique used to predict how a ligand binds to a target protein.
More specifically, it aims to determine:
- the binding pose (orientation and position)
- an estimate of binding affinity
Docking works by:
- generating multiple possible ligand conformations
- placing them into the protein binding site
- scoring each pose using mathematical functions
It is widely used because:
- it is fast (seconds to minutes per compound)
- it can screen large compound libraries
- it provides a first approximation of binding interactions
This makes docking an ideal tool for:
- virtual screening
- hit identification
- early-stage filtering
In fact, docking remains one of the most commonly used methods in structure-based drug design.
The Strengths of Docking
Docking is powerful because it allows researchers to:
1. Screen at Scale
Evaluate thousands to millions of compounds efficiently.
2. Identify Binding Modes
Predict how a molecule fits into the active site.
3. Prioritize Candidates
Rank molecules based on predicted affinity.
The Limitations of Docking
Despite its utility, docking has inherent limitations.
1. It Treats Systems as Static
Docking typically assumes that the protein structure is rigid or only minimally flexible.
But in reality:
- proteins are dynamic
- binding sites change shape
- interactions evolve over time
This simplification can lead to inaccurate predictions.
2. It Relies on Simplified Scoring Functions
Docking scores are approximations.
They often fail to fully capture:
- solvent effects
- entropy contributions
- long-range interactions
As a result, docking can produce false positives or mis-rank compounds.
3. It Cannot Capture Time-Dependent Behavior
Docking provides a snapshot.
But binding is a process, not a single moment.
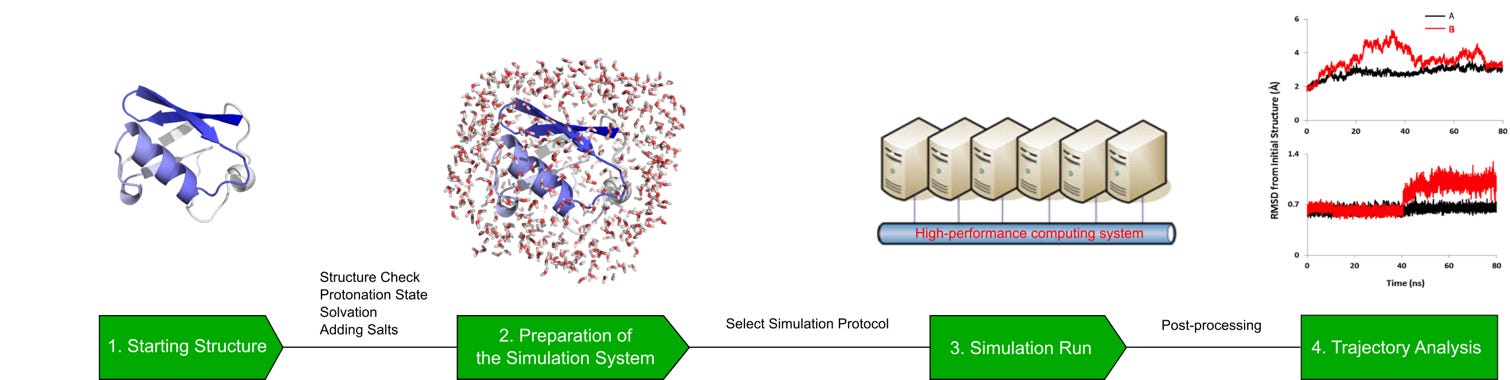
What Is Molecular Dynamics (MD)?
Molecular dynamics simulations take a fundamentally different approach.
Instead of predicting a static pose, MD simulates:
how atoms and molecules move over time.
By solving Newton’s equations of motion, MD tracks:
- atomic trajectories
- conformational changes
- interaction stability
over nanoseconds to microseconds.
This allows researchers to observe:
- how a ligand behaves inside a binding pocket
- whether interactions are stable
- how the protein structure adapts
The Strengths of Molecular Dynamics
1. Capturing Molecular Motion
MD reveals that proteins are not rigid structures.
They continuously fluctuate and adapt.
This dynamic behavior is critical for understanding binding.
2. Evaluating Stability
A molecule that docks well may not remain bound.
MD helps answer:
Does the ligand stay in place over time?
3. Incorporating Realistic Conditions
MD simulations can model:
- solvent (water molecules)
- temperature
- pressure
This makes the system closer to biological reality.
4. Revealing Hidden Binding Sites
MD can uncover:
- transient pockets
- conformational changes
that are not visible in static structures.
The Limitations of Molecular Dynamics
While MD provides deeper insights, it comes with trade-offs.
1. Computational Cost
MD simulations are significantly more expensive than docking.
They require:
- high-performance computing
- longer runtimes
2. Complexity
Setting up accurate MD simulations requires:
- correct force fields
- proper system preparation
- careful interpretation
3. Timescale Constraints
Even with modern computing, capturing very long biological processes remains challenging.
Docking vs Molecular Dynamics: The Core Difference
At a high level, the distinction can be summarized as:
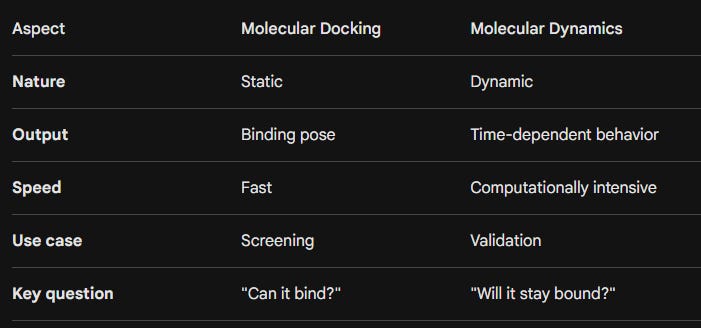
Docking predicts possibility.
MD evaluates reality.
Why Docking Alone Is Not Enough
In many workflows, docking is used as the primary decision-making tool.
But this creates a gap.
A molecule may:
- score well in docking
- appear to bind strongly
but fail when:
- protein flexibility is considered
- solvent effects are included
- dynamics are simulated
This is one of the key reasons why many early-stage predictions do not translate into successful candidates.
The Power of Combining Docking and MD
Rather than choosing one over the other, the most effective approach is integration.
A typical workflow looks like:
Docking
- screen large libraries
- identify promising candidates
Molecular Dynamics
- validate binding stability
- analyze interactions over time
Advanced Methods (e.g., FEP)
- quantify binding free energy
- refine ranking
This combination leverages:
- the speed of docking
- the accuracy of MD
and results in better-informed decisions.
Medvolt’s Approach: From Prediction to Validation
At Medvolt, we treat docking and molecular dynamics not as separate tools, but as connected stages of a unified workflow.
In practical terms, this means:
- docking is used for rapid exploration
- MD is used for dynamic validation
- physics-based methods refine predictions further
Our workflows integrate:
- structure-aware docking with AI-based rescoring
- molecular dynamics simulations for stability analysis
- free energy calculations for accurate affinity prediction
This allows us to move beyond:
“Does it bind?”
to:
“Does it bind, remain stable, and behave correctly under realistic conditions?”
Why This Matters for Drug Discovery
The distinction between docking and MD has real implications.
1. Better Candidate Selection
Combining both methods reduces false positives.
2. Lower Failure Rates
Early-stage validation helps avoid costly downstream failures.
3. Improved Understanding of Mechanism
Dynamic simulations provide insights into:
- binding pathways
- conformational changes
- interaction networks
4. More Efficient Pipelines
By filtering candidates more intelligently, resources can be focused on the most promising molecules.
The Bigger Picture: From Approximation to Reality
Drug discovery is moving toward more realistic modeling of biological systems.
This means:
- moving beyond static representations
- incorporating time and physics
- integrating multiple computational approaches
Docking remains essential.
But it is no longer sufficient on its own.
Conclusion
Molecular docking and molecular dynamics are not competing techniques.
They are complementary.
Docking provides speed and scale.
Molecular dynamics provides depth and realism.
Together, they form a more complete picture of molecular interactions.
As drug discovery becomes more complex, relying on a single method is no longer enough.
The future lies in integrated, physics-aware workflows that combine prediction with validation.
Because in the end, the goal is not just to predict binding.
It is to understand it.
