



What Really Happens During Molecular Binding
Molecular binding is not static but a dynamic, multi-step process shaped by motion, solvent, and thermodynamics, critical for real-world drug discovery.
Introduction
In drug discovery, one of the most fundamental questions is:
How does a molecule bind to its target protein?
At first glance, the answer seems straightforward.
A molecule fits into a binding site.
Interactions form.
Binding occurs.
This intuitive picture is often described as the “lock-and-key” model, where the protein is the lock and the ligand is the key.
But this model, while useful, is incomplete.
In reality, molecular binding is not a static event.
It is a dynamic, multi-step process shaped by motion, environment, and thermodynamics.
Understanding this process is critical for designing molecules that not only bind, but bind effectively under real biological conditions.
The Simplified View: Lock-and-Key
The lock-and-key model assumes:
- the protein has a fixed structure
- the ligand fits perfectly into the binding site
- binding occurs without structural changes
This model has been historically important in explaining molecular recognition.
But it fails to capture the complexity of real biological systems.
Proteins are not rigid locks.
And ligands are not perfectly shaped keys waiting to be inserted.
In practice, even initial computational predictions such as docking, while useful for identifying plausible binding poses, operate within this simplified framework and must be interpreted carefully.
The Reality: A Dynamic Interaction
In reality, both proteins and ligands are constantly in motion.
At physiological temperatures:
- atoms vibrate
- side chains rotate
- binding pockets fluctuate
This means that binding is not a simple fit.
It is a process of adaptation.
Two key models better describe this behavior:
1. Induced Fit
In induced fit, the protein changes shape in response to the ligand.
- the ligand approaches the binding site
- the protein adjusts its conformation
- a stable complex forms
2. Conformational Selection
In conformational selection, the protein already exists in multiple conformations.
- the ligand binds to one of these pre-existing states
- the equilibrium shifts toward the bound conformation
This highlights an important idea:
The binding site is not a single structure, but a distribution of states.
Capturing this distribution computationally requires approaches that go beyond static modeling and incorporate structural flexibility explicitly.
Step-by-Step: What Happens During Binding
Binding is a sequence of events rather than a single moment.
1. Diffusion and Encounter
The ligand diffuses through the solvent environment.
- random motion brings it close to the protein
- long-range electrostatic interactions may guide it
2. Initial Contact
The ligand forms weak, transient interactions with the protein surface.
- hydrogen bonds
- electrostatic interactions
- van der Waals contacts
At this stage, binding is unstable and reversible.
3. Orientation and Adjustment
The ligand reorients itself within or near the binding pocket.
Simultaneously:
- protein residues shift
- side chains adapt
- water molecules rearrange
This is often where many computational predictions diverge from reality.
A pose that appears favorable in docking may not sustain itself once these dynamic adjustments begin.
4. Stabilization
If favorable interactions are established:
- stronger contacts form
- the ligand becomes more tightly bound
- the system moves toward a lower energy state
5. Dynamic Equilibrium
Even after binding, the system is not static.
- interactions fluctuate
- the ligand may shift within the pocket
- partial unbinding events can occur
Binding is better understood as an ensemble of states, not a fixed configuration.
The Hidden Role of Water
One of the most overlooked aspects of molecular binding is the role of water.
In biological systems:
- binding sites are often hydrated
- water molecules form structured interaction networks
During binding:
- some water molecules are displaced
- others remain and mediate interactions
These effects can significantly influence binding affinity and specificity.
In computational workflows, accurately capturing water behavior often requires molecular dynamics simulations, where solvent molecules are explicitly modeled rather than approximated.
The Role of Thermodynamics
Binding is governed by thermodynamics, particularly free energy (ΔG).
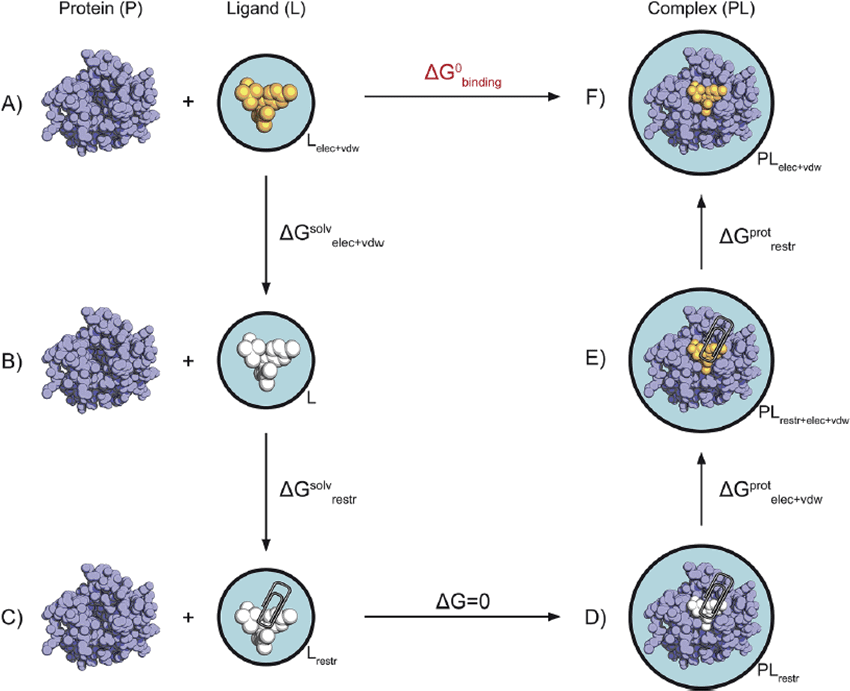
Two key components influence binding:
1. Enthalpy (ΔH)
Represents interaction strength:
- hydrogen bonds
- electrostatic forces
- van der Waals interactions
2. Entropy (ΔS)
Represents changes in disorder:
- loss of ligand flexibility
- conformational constraints
- solvent rearrangement
Binding occurs when:
ΔG = ΔH – TΔS is negative
This balance explains why strong interactions alone are not sufficient.
A molecule must also behave favorably within its environment.
Quantifying this balance accurately often requires physics-based approaches such as free energy calculations, which go beyond heuristic scoring methods.
Why Static Models Fall Short
Traditional approaches often rely on static representations.
For example:
- docking predicts a single binding pose
- scoring functions estimate affinity using simplified models
While useful for early-stage filtering, these methods do not capture:
- conformational flexibility
- time-dependent interactions
- solvent dynamics
This is one of the reasons why many compounds that appear promising computationally fail during experimental validation.
Capturing Reality: From Prediction to Simulation
To address these limitations, modern workflows increasingly incorporate molecular dynamics (MD) simulations.
MD enables researchers to:
- simulate atomic motion over time
- observe ligand stability within the binding pocket
- analyze interaction persistence
- capture protein flexibility and solvent effects
This transforms binding from a static prediction into a time-resolved process.
In practical applications, combining docking with MD allows for:
- rapid exploration of candidate molecules
- followed by dynamic validation under realistic conditions
Integrating AI and Physics in Binding Analysis
Recent advances in AI have significantly improved:
- structure prediction
- molecule generation
- large-scale pattern recognition
However, molecular binding is ultimately governed by physical laws.
As a result, the most effective approaches combine:
- AI-driven predictions
- structure-based modeling
- molecular dynamics simulations
- thermodynamic calculations
In integrated pipelines, AI can guide where to search, while physics-based simulations determine what is actually viable.
This hybrid approach is increasingly being adopted to improve both accuracy and efficiency in drug discovery workflows.
A Practical Perspective: How Binding Is Studied Today
In modern computational pipelines, understanding binding typically involves multiple stages:
- Docking: identify plausible binding poses
- Molecular Dynamics: evaluate stability and interaction dynamics
- Free Energy Methods (e.g., FEP): quantify binding affinity with higher accuracy
Such multi-layered workflows help reduce uncertainty and improve confidence in selected candidates.
In applied settings, integrating these steps into a unified pipeline allows researchers to move beyond isolated predictions toward a more consistent understanding of molecular behavior.
What This Means for Drug Discovery
A deeper understanding of molecular binding has important implications:
- Better Molecule Design: Design strategies that account for dynamics and thermodynamics improve success rates.
- Reduced Late-Stage Failures: Early identification of unstable interactions prevents costly downstream failures.
- More Efficient Optimization: Mechanistic insights guide targeted modifications rather than blind iteration.
- Improved Biological Relevance: Simulating realistic environments leads to more reliable predictions.
Conclusion
Molecular binding is far more complex than the traditional lock-and-key analogy suggests.
It is a dynamic, multi-step process influenced by:
- protein flexibility
- ligand motion
- solvent interactions
- thermodynamic balance
Understanding this complexity is essential for designing effective drug candidates.
As computational methods evolve, the focus is shifting from:
predicting whether a molecule binds
to:
understanding how it binds, how stable it is, and how it behaves over time.
In this context, approaches that integrate structure-based modeling, dynamic simulations, and thermodynamic analysis provide a more complete and reliable picture of molecular interactions.
Because in the end, successful drug discovery is not about finding molecules that simply fit.
It is about finding molecules that work within the dynamic reality of biological systems.
