



Why Most Binding Predictions Fail in Drug Discovery
Binding predictions often fail due to oversimplified models that ignore dynamics, solvent effects, and thermodynamics, creating a gap between computation and reality.
Introduction
Predicting how a molecule binds to a protein is one of the most critical steps in drug discovery.
It is also one of the most misunderstood.
At first glance, the process seems straightforward:
- identify a target protein
- dock a molecule into its binding site
- calculate a score
- select the best candidates
But in practice, this approach often fails.
A molecule that looks promising computationally may:
- fail to bind experimentally
- bind weakly or transiently
- lose activity in biological systems
This gap between prediction and reality is one of the key reasons why drug discovery remains slow, expensive, and uncertain.
So why do most binding predictions fail?
The answer lies in how we model reality.
The Illusion of Static Binding
One of the biggest limitations in binding prediction is the assumption of static systems.
Most traditional methods, especially molecular docking, treat:
- proteins as rigid structures
- ligands as fixed conformations
- interactions as instantaneous
But biological systems do not behave this way.
Proteins:
- undergo conformational changes
- exhibit flexibility in binding sites
- adapt to ligand presence
Ligands:
- rotate, bend, and reorient
- interact dynamically with the environment
By reducing this complexity to a single snapshot, many prediction methods miss the true nature of binding.
The Scoring Function Problem
Docking relies heavily on scoring functions to estimate binding affinity.
These functions approximate:
- hydrogen bonding
- electrostatic interactions
- van der Waals forces
However, they often fail to capture:
- entropy effects
- solvent interactions
- long-range electrostatics
- dynamic rearrangements
As a result:
- false positives are common
- weak binders may be overestimated
- strong candidates may be overlooked
This is not a flaw of docking itself, but a limitation of simplified models.
Ignoring Protein Flexibility
Binding sites are not static cavities.
They:
- expand and contract
- expose hidden pockets
- change shape upon ligand binding
Many failures occur because:
- the docking pose fits a rigid structure
- but does not align with the protein’s dynamic behavior
In reality, binding often involves:
- induced fit
- conformational selection
Without accounting for these mechanisms, predictions remain incomplete.
The Hidden Complexity of Solvent
Water is not just a background medium.
It actively participates in molecular binding.
Water molecules can:
- stabilize interactions
- form hydrogen bond bridges
- be displaced during binding
Ignoring solvent effects can lead to:
- incorrect affinity estimates
- misinterpretation of binding modes
Capturing these effects requires explicit modeling, which is often absent in basic prediction workflows.
Entropy: The Missing Piece
Binding is governed by free energy (ΔG), which includes:
- enthalpy (interaction strength)
- entropy (system disorder)
Many computational methods focus on enthalpy while neglecting entropy.
But entropy plays a crucial role:
- restricting ligand motion reduces entropy
- rearranging solvent affects system energy
- protein flexibility contributes to overall balance
Failing to account for entropy leads to inaccurate predictions, even when interactions appear strong.
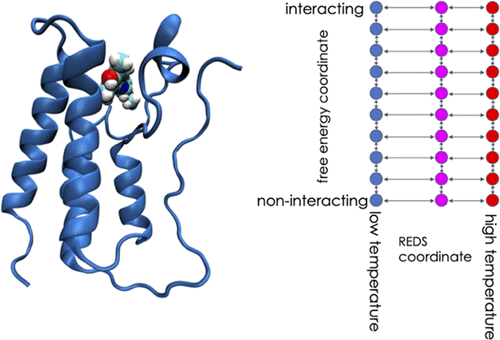
Overreliance on Single Methods
Another major reason for failure is reliance on a single computational approach.
For example:
- using docking alone for decision-making
- selecting top-ranked molecules without validation
This creates a pipeline where:
- early errors propagate forward
- weak candidates are prioritized
- experimental resources are wasted
Binding prediction is not a one-step problem.
It requires multiple layers of validation.
The Gap Between Prediction and Reality
Taken together, these limitations create a significant gap:
Predicted binding ≠ Real binding behavior
This gap is responsible for:
- high false positive rates
- poor reproducibility
- late-stage failures
Closing this gap requires a shift in how binding is evaluated.
Moving Toward Dynamic, Physics-Based Understanding
To improve accuracy, modern drug discovery is moving toward physics-based and dynamic modeling approaches.
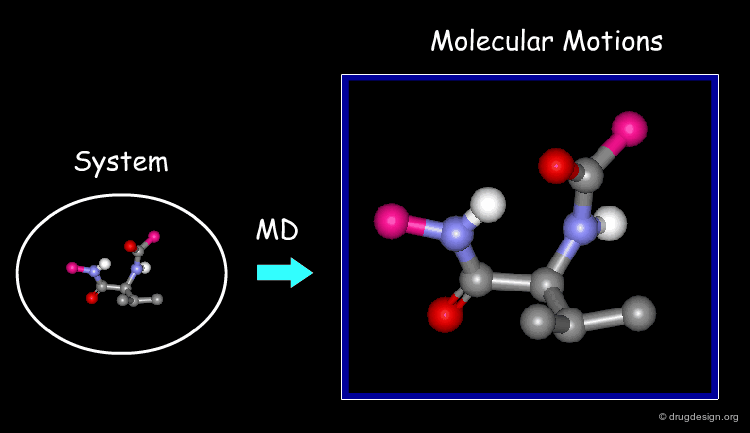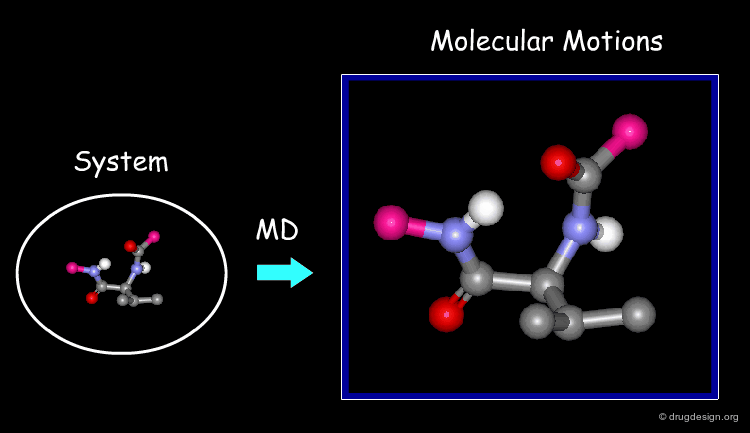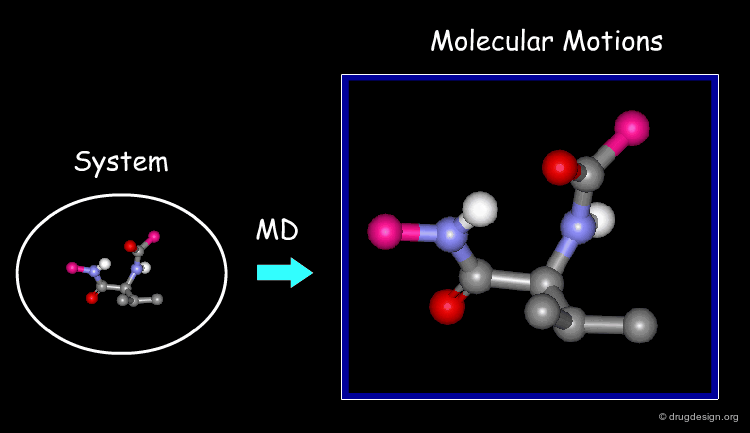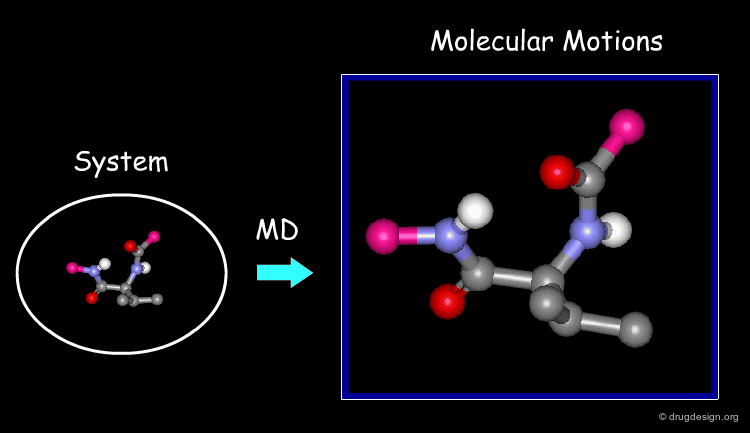
Molecular Dynamics (MD)
MD simulations capture:
- protein flexibility
- ligand movement
- time-dependent interactions
They answer a critical question:
Does the ligand remain stable over time?
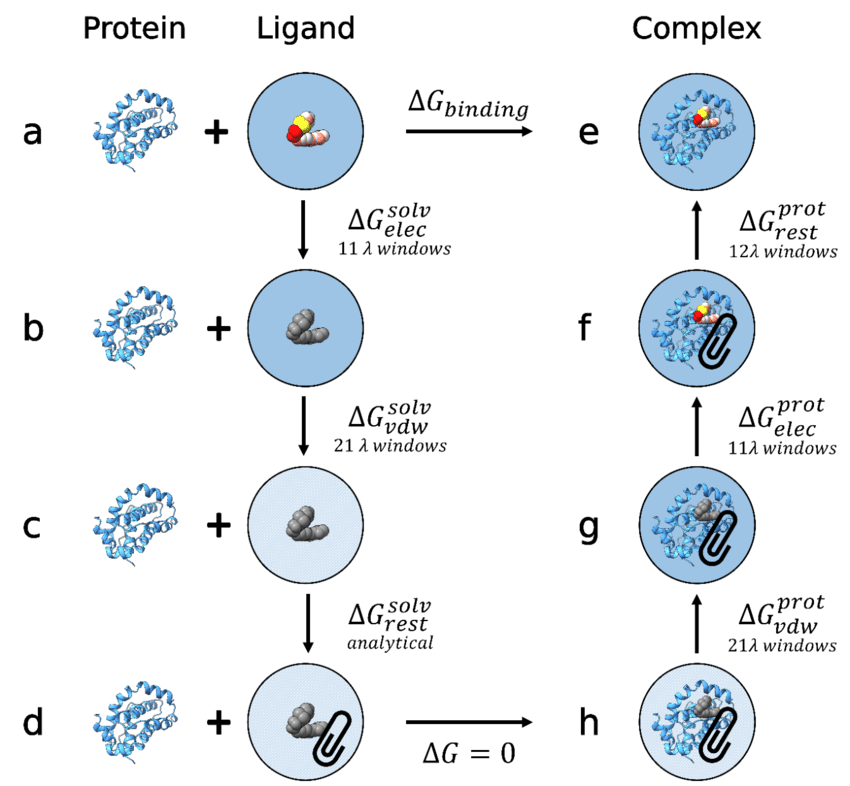
Free Energy Calculations (FEP)
FEP methods estimate binding affinity more accurately by:
- incorporating thermodynamics
- accounting for solvent and entropy
- modeling realistic interactions
These methods provide quantitative insights rather than heuristic scores.
Integrated Workflows
The most effective pipelines combine:
- Docking → fast exploration
- MD → dynamic validation
- FEP → precise quantification
This layered approach reduces uncertainty and improves decision quality.
Medvolt’s Approach: Reducing Failure Through Integration
At Medvolt, we address binding prediction as a multi-dimensional problem, not a single calculation.
Our workflows are designed to bridge the gap between prediction and reality.
Structure-Aware Docking
We use docking as an initial step, but enhance it with:
- AI-based rescoring
- structural context awareness
- better prioritization of candidates
This improves early-stage filtering.
Molecular Dynamics for Validation
Promising candidates are evaluated using MD simulations to:
- assess stability
- analyze interaction persistence
- capture conformational changes
This ensures that selected molecules behave realistically.
Physics-Based Free Energy Calculations
We apply FEP and related methods to:
- quantify binding affinity
- refine rankings
- reduce false positives
This adds a layer of decision-grade accuracy.
Multi-Parameter Optimization
Binding is not evaluated in isolation.
We simultaneously consider:
- potency
- selectivity
- ADMET properties
- developability
This reduces downstream risk.
Integrated AI + Physics Framework
Our platform combines:
- generative AI for molecule design
- knowledge-driven insights
- physics-based simulations
This ensures that predictions are:
- data-informed
- physically meaningful
- experimentally relevant
Why This Matters
Improving binding prediction has direct impact on drug discovery outcomes.
- Better Candidate Selection: More accurate predictions lead to higher-quality molecules entering the pipeline.
- Reduced Experimental Burden: Fewer false positives mean fewer unnecessary experiments.
- Lower Failure Rates: Early validation reduces costly late-stage failures.
- Faster Development: Better decisions accelerate the overall discovery process.
The Future of Binding Prediction
The field is moving toward:
- dynamic modeling
- integrated pipelines
- AI + physics hybrid approaches
The goal is not just to predict binding.
It is to understand it in realistic conditions.
This requires:
- better models
- better data integration
- better validation frameworks
Conclusion
Most binding predictions fail not because the idea is flawed, but because the models are incomplete.
Static assumptions, simplified scoring, and lack of validation create a gap between prediction and reality.
Closing this gap requires:
- embracing molecular dynamics
- incorporating thermodynamics
- integrating multiple computational approaches
At Medvolt, this philosophy is central to how we approach drug discovery.
Because in the end, success is not about finding molecules that appear to bind.
It is about identifying molecules that truly work in the complex, dynamic environment of biology.
